近年来,重组蛋白应用有了显著增长,与此同时,用于表达和纯化的重组蛋白技术和产品的应用也相应得以增长。用标签融合于重组蛋白中,进而方便重组蛋白的纯化和检验,这一优势已被广泛认可。
一个蛋白质表达系统包括:合适的启动子、其他调控序列、编码所需重组蛋白的基因的载体,以及一些其他的组成部分。用于表达的载体都有商品化的产品,这些商品化的载体被设计成带有适合特定宿主(如大肠杆菌或哺乳动物细胞)的调控区域,它们所带有的抗性标记基因使得选择正确的克隆变得简单易行。
宿主的选择
宿主的选择不仅影响蛋白的表达,而且也影响产品的随后纯化。许多宿主表达系统都可以使用,其中包括细菌、昆虫、哺乳动物细胞等。每种宿主系统都有它的优缺点。如表1所示。

表1
载体的选择
载体家族的选择主要受限于宿主的选择。一旦宿主被确定,有许多不同的载体可供选择,比如从简单的表达载体到含有特定序列从而能使重组蛋白分泌出去的载体。

常见真核表达启动子和原核表达载体启动子如表2所示。

表2
标签的选择
亲和层析标签分为大标签和小标签,大标签如GST、MBP,通常为几十KD,大标签一般在纯化后需要切除。通常,大标签可能也会提高表达蛋白的可溶性。小标签多为6-10个氨基酸组成,如His,Strep(II),Flag等,且在纯化后不需要对标签进行切除。最常用的两种标签是六组氨酸标签和GST标签。

原核表达载体构建
引物设计
目的基因CDS区域克隆
目的基因酶切
质粒进行酶切线性化、琼脂糖凝胶电泳、胶回收纯化
目的基因和线性化质粒连接
转化到大肠杆菌DHa中
鉴定(PCR、测序、酶切)
阳性菌落接种、诱导、扩大培养
1、NCBI查找目的基因的CDS序列
打开NCBI网站(https://www.ncbi.nlm.nih.gov/),在下拉框选择Gene,输入目的蛋白名称;

找到对应物种,也可以选择右侧框中的物种查找;

页面往下拉,找到mRNA and Protein(s),选择正确的isoform,NMxxx表示mRNA序列,NPxxx表示蛋白质序列;
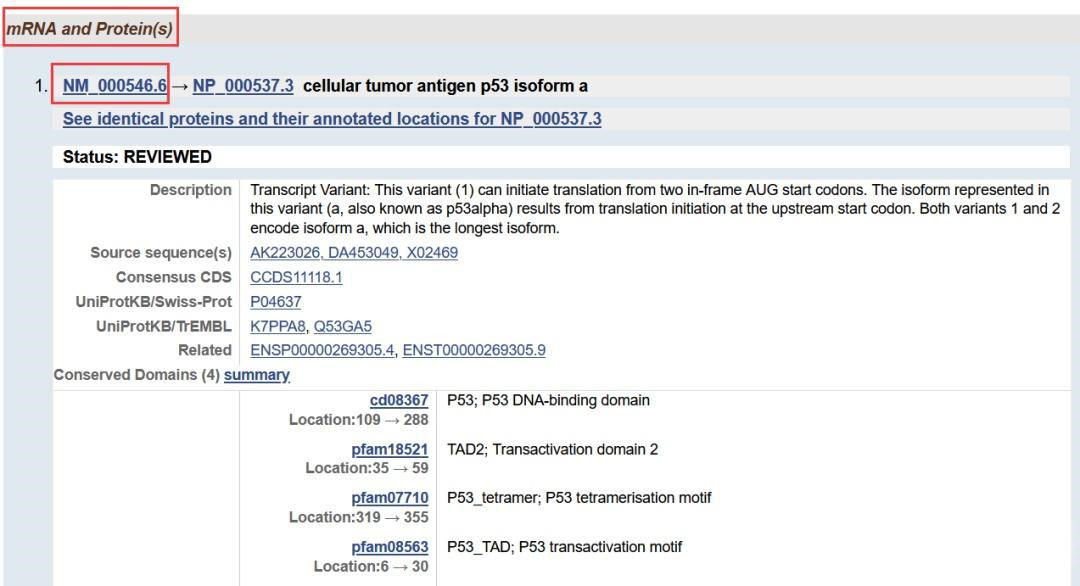
新页面往下拉,点击CDS,点击右侧FASTA,即可得到目的基因的mRNA序列,复制粘贴到新的文本,以备使用。

2、选择合适的表达载体,确定双酶切位点
常用原核表达载体如pET15b,pET28a,pGEX4T1,pGEX-6p-1等。如图所示,pET15b常用的酶切位点如:Nde I、Xho l、BamH I。

双酶切位点的选择原则:
1)酶切位点存在于载体上,不存在于基因序列中;
2)两个酶有共同的缓冲体系;
3)两个酶在载体上酶切位点位置不能太近。
3、Primer 5预测目的序列酶切位点
1.打开Primer 5,选择file - new -DNAsequence,输入上述保存的CDS序列,点击As Is;
2.点击左侧Enzyme框,看一下载体上选择的双酶切位点是否在右侧框中,如果不在,从左侧框中添加到右侧框中;
3.点击OK即可查看CDS序列中有无选定的酶切位点,如果选定的双酶切位点存在于目的蛋白的CDS序列中,需重新选择双酶切位点。
4、双酶切buffer
因为我们采用的是双酶切,那么要求所用的两个酶都必须有相同的酶切buffer ,这样才能在同一缓冲液中同时进行酶切。酶切buffer可以参考产品说明书确定。
5、目的基因CDS序列引物设计
从细胞中提取mRNA→逆转录成cDNA→扩增目的片段的CDS序列。
引物设计:扩增目的基因CDS全序列,选取序列前端20-25个碱基作为上游引物,末端20-25个碱基的反向互补序列作为下游引物,这样上述P53序列上下游引物分别为:
F:ATGGAGGAGCCGCAGTCAG;
R:TCAGTCTGAGTCAGGCCCTTC。
如若扩增片段序列,需要在上游引物加上起始密码子,在下游引物加上终止密码子。常见的起始密码子为ATG,终止密码子通常为TGA,TAA,TAG。若载体上无标签,构建载体时可在上游引物前端或下游引物后端添加标签序列,这样即可把标签添加到序列的N端或C端。
引物设计的原则:
1)引物长度一般为15-30bp,常用的为18-27bp,但不能大于38bp;
2)引物GC含量一般为40%-60%,以45-55%为宜,上下游引物GC含量和Tm值要保持接近;
3)引物所对应的模板序列的Tm值最好在72℃左右;
4)3'端最好不要是连续碱基,GGG或CCC会导致错误的引发,同时3'端最后一个碱基最好不要是A或T,否则容易导致错配;
5)以公式Tm=4*(G+C)+2*(A+T)-5计算Tm值,也就是退火温度。选择较低Tm值的引物的退火温度为反应的退火温度,最好保证每个引物的Tm值相匹配,且在70-75℃范围内;
6)在DNA测序和PCR中最好用5'末端稳定(GC含量多),而3'端不稳定(AT含量多)的引物,这种引物的结构可以有效地消除假引发反应;
7)引物和产物之间的Tm值相差别太大,20摄氏度范围内最好。
引物特异性判断:在NCBI数据库中检测引物的特异性。
1)NCBI主页最下端找到Primer-BLAST,打开;

2)输入上下游引物序列,点击最下方【Get Primers】;

3)可以发现,页面中100%匹配的基因序列只有P53,说明引物的特异性较好;

4)上下游引物各自加入酶切位点。注意:上游引物加入酶切位点时容易引起移码突变,所以要保证在起始密码子前端加入的是三个碱基倍数的读码框,必要时需要人为加入1-2个碱基数,但注意避开终止密码子。下游引物因为酶切位点前存在终止密码,因此不需要考虑酶切位点引入之后读码框发生的变化。
6、转化:将质粒 DNA 导入细菌的过程称为转化。
在自然条件下,很多质粒都可通过细菌接合作用转移到新的宿主内,但在人工构建的质粒载体中,一般缺乏此种转移所必需的mob基因,因此不能自行完成从一个细胞到另一个细胞的接合转移。如需将质粒载体转移进受体细菌,需诱导受体细菌产生一种短暂的感受态以摄取外源DNA,常用的方法有CaCl2法和电转法。
转化的一般过程:
1)取出感受态大肠杆菌,立即放于冰上,使其慢慢融化;
2)将16ul连接产物加入100ul感受态大肠杆菌中,轻轻混匀,冰上孵育30分钟;
3)将EP管放入42℃水浴箱中热激90秒,再迅速放于冰上2分钟;
4)EP管中加入400u LB培养基,置于摇床上,37℃,500rpm培养45分钟;
5)500rpm离心1-2分钟,留100ul培养基上清,将转化后的大肠杆菌轻轻混匀,涂于含抗生素的LB固体培养基上,37℃恒温培养箱中倒置培养过夜。
7、双酶切及鉴定
挑取单菌落,接种,培养,取100ul菌液送测序;其余菌液提质粒,双酶切PCR,琼脂糖凝胶鉴定。
8、测序正确的阳性菌落即可扩大培养,用于后期蛋白纯化研究。

